
アルポート症候群
難聴や視力障害を伴う遺伝性の糸球体腎炎です。糸球体基底膜(GBM)にあるⅣ型コラーゲンの遺伝子変異が原因で起こります。X染色体性の遺伝のため、男性に多く、幼少期から顕微鏡的血尿が認められ、30歳代までに透析が必要になります。
原因は ?
糸球体基底膜(GBM)の緻密層の主な構成成分であるⅣ型コラーゲンのα鎖の遺伝子変異が原因です。
遺伝型式は ?
X染色体優性遺伝(85%),常染色体劣性遺伝(10%),常染色体優性遺伝に分類されます。 X染色体性では、保因者(2つのX染色体の一方に遺伝子変異を認めます)の母親から子供へ2分の1の確率で遺伝します。遺伝子変異を受け継ぐと、X染色体を1つしか持たない男の子は早期から発症します。それに対して、女性は2本のX染色体を持つため、軽症です。ただし、稀ですが、女性患者でも透析が必要になる場合もあるので、注意が必要です。
症状・検査所見・経過・予後は ?
腎障害だけでなく、聴力障害や眼症状を伴うことが特徴です。ただし、同時に認められない場合もあります。
腎障害
多くは生後間もない頃から顕微鏡的血尿が認められます。加齢とともに蛋白尿も加わり、ネフローゼ症候群を呈することもあります。X染色体性の男性患者は発症も早く進行性に腎機能が低下し,多くは30歳代までに末期腎不全に至ります.X染色体性の女性患者の腎機能は比較的保たれ,多くは軽度の血尿,蛋白尿のみを認めます。
聴力障害
30~40%に両側性進行性の感音性難聴を認めます。
眼症状
円錐水晶体,水晶体脱臼,皮質部白内障などを認めます。
診断は ?
家族歴と腎生検により診断されます。
診断基準
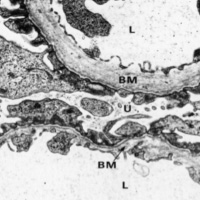
- 尿検査異常または腎臓病の家族歴
- 基底膜の特徴的な電顕所見(図)
- 進行性の高音性感音性難聴
- 眼症状 のうち3つを満たすものをアルポート症候群と診断します。
3.4.を伴わない患者でも特徴的な電顕所見や免疫組織学的解析により診断できます。 またX染色体性遺伝であれば皮膚生検でのα5鎖の染色でも診断可能です。 ただし、20歳未満の女性で腎機能障害を示す場合は常染色体劣性遺伝を疑います。
鑑別診断は ?
難聴などの腎外症状がない場合は、腎生検によって糸球体基底膜菲薄化症候群やIgA腎症を鑑別する必要があります。
治療は ?
根本的治療はないため、腎機能障害には腎機能保護を目的とした保存的治療を行います。腎移植も行われるが,まれに移植後抗基底膜抗体糸球体腎炎を発症する(3~5%)ので.注意が必要です。